
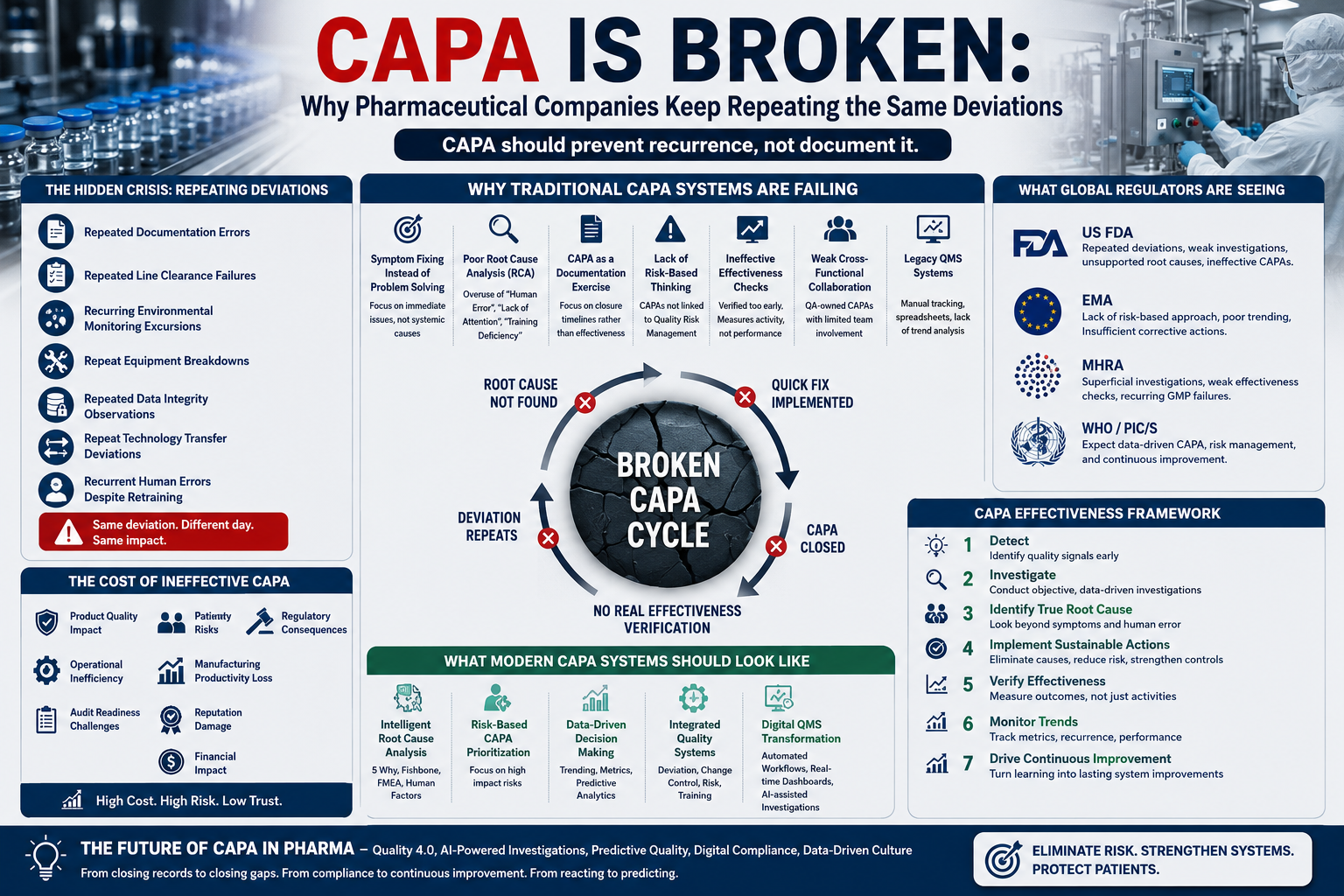
Introduction
In the pharmaceutical industry, few quality processes are as critical—or as misunderstood—as Corrective and Preventive Action (CAPA).
For decades, CAPA has been regarded as the backbone of Pharmaceutical Quality Systems (PQS), serving as the primary mechanism for identifying problems, investigating root causes, implementing corrective actions, preventing recurrence, and driving continuous improvement.
Regulatory agencies worldwide—including the US FDA, EMA, MHRA, WHO, and PIC/S—expect pharmaceutical organizations to maintain robust CAPA systems capable of transforming quality events into learning opportunities and sustainable improvements.
Yet despite significant investments in:
- Quality Management Systems (QMS)
- Electronic Quality Systems (eQMS)
- GMP Training Programs
- Internal Audits
- Regulatory Compliance Initiatives
- Investigation Processes
- Quality Risk Management Programs
many pharmaceutical companies continue to experience the same deviations repeatedly.
The industry faces a troubling reality:
A deviation is investigated.
A CAPA is opened.
Actions are implemented.
The CAPA is closed.
Six months later, the same issue returns.
The problem is not that organizations lack CAPA systems.
The problem is that many organizations have become CAPA-compliant rather than CAPA-effective.
Today, recurring deviations, repeat audit observations, repeated Out-of-Specification (OOS) results, recurring environmental monitoring excursions, repeat customer complaints, and recurring regulatory findings indicate a deeper systemic issue:
Traditional CAPA systems are failing modern pharmaceutical operations.
The Hidden Crisis: Repeating Deviations Across the Industry
Almost every pharmaceutical facility experiences recurring quality events.
While individual deviations may appear isolated, industry-wide patterns reveal a more concerning trend.
Repeated Documentation Errors
Organizations continue to observe:
- Missing entries in batch records
- Incorrect data recording
- Incomplete logbooks
- GDP non-compliance
- Missing signatures
Despite repeated retraining efforts, the same errors reappear.
The issue often extends beyond employee behavior and points toward poor process design, excessive documentation burden, inadequate supervision, or ineffective workflows.
Repeated Line Clearance Failures
Manufacturing facilities frequently encounter:
- Incorrect status labels
- Residual materials
- Cleaning verification failures
- Product mix-up risks
Investigations often conclude:
“Operator failed to follow procedure.”
However, repeated occurrences suggest weaknesses in process design, visual management systems, workplace organization, and operational controls.
Recurring Environmental Monitoring Excursions
Microbiology teams repeatedly investigate:
- Elevated viable counts
- Non-viable particle excursions
- Water system contamination
- Cleanroom environmental deviations
Organizations often implement localized corrections while failing to address broader contamination control strategies.
Repeat Equipment Breakdowns
Engineering departments commonly experience recurring failures involving:
- HVAC systems
- Water systems
- Compression equipment
- Packaging machinery
- Production assets
Replacing components without addressing lifecycle management, preventive maintenance effectiveness, and reliability engineering often results in recurring failures.
Repeated Data Integrity Observations
Many inspections continue to identify:
- Incomplete audit trail reviews
- Unauthorized data changes
- Poor documentation practices
- Inadequate electronic record controls
Repeated training alone rarely resolves systemic data governance deficiencies.
Repeat Technology Transfer Deviations
During technology transfer projects, organizations frequently experience:
- Process variability
- Documentation inconsistencies
- Analytical method issues
- Validation gaps
Repeated events often indicate weak knowledge transfer processes rather than isolated execution failures.
Recurrent Human Errors Despite Retraining
Perhaps the most common recurring CAPA observation involves repeated human errors.
Employees are retrained.
Training records are completed.
CAPAs are closed.
The same deviations return.
At this point, training is not the solution.
The system is.
Why Traditional CAPA Systems Are Failing
A. Symptom Fixing Instead of Problem Solving
One of the most significant weaknesses in CAPA management is the tendency to address symptoms rather than causes.
Consider a repeated documentation error.
The investigation concludes:
“Operator entered incorrect data.”
Corrective action:
“Retrain operator.”
CAPA closed.
However, critical questions remain unanswered:
- Why was the error possible?
- Why was it not detected sooner?
- Why did existing controls fail?
- Could another operator make the same mistake?
Organizations often solve the immediate problem while leaving systemic vulnerabilities intact.
This creates a cycle of recurring deviations disguised as resolved issues.
B. Poor Root Cause Analysis (RCA)
Root Cause Analysis remains one of the weakest aspects of pharmaceutical investigations.
Frequently cited root causes include:
- Human Error
- Operator Negligence
- Lack of Attention
- Failure to Follow Procedure
- Training Deficiency
These are rarely true root causes.
Instead, they are often the final visible manifestation of deeper system weaknesses.
For example:
An operator skips a procedural step.
The investigation concludes:
“Human Error.”
But deeper analysis may reveal:
- Excessive workload
- Poor procedure design
- Inadequate visual controls
- Fatigue
- Poor ergonomics
- Confusing instructions
- Ineffective supervision
Human error is often the outcome of a flawed system—not the root cause itself.
C. CAPA as a Documentation Exercise
In many organizations, CAPA has become a paperwork activity rather than an improvement process.
Success is often measured by:
- CAPA closure rates
- Investigation turnaround time
- On-time completion metrics
As a result:
Teams prioritize closure over effectiveness.
The objective becomes:
“Close the CAPA.”
Instead of:
“Eliminate the risk.”
This mindset creates a dangerous compliance illusion.
Records appear complete.
Problems remain unresolved.
D. Lack of Risk-Based Thinking
Modern pharmaceutical operations are increasingly complex.
Not all deviations carry equal risk.
However, many CAPA systems treat every issue similarly.
Without integrating Quality Risk Management (QRM), organizations struggle to:
- Prioritize critical issues
- Allocate resources effectively
- Focus on patient-impacting risks
- Prevent significant quality failures
A risk-blind CAPA system inevitably becomes overloaded and ineffective.
E. Ineffective Effectiveness Checks
One of the most common CAPA failures involves weak effectiveness verification.
Typical effectiveness checks include:
- Confirmation that training occurred
- Verification that SOP revisions were issued
- Evidence that meetings were conducted
These activities confirm completion.
They do not confirm effectiveness.
True effectiveness should evaluate:
- Has recurrence decreased?
- Has process capability improved?
- Have quality metrics improved?
- Has risk been reduced?
Many organizations verify activity completion instead of performance improvement.
F. Weak Cross-Functional Collaboration
CAPA effectiveness requires cross-functional ownership.
Yet many CAPAs remain largely QA-driven.
Production, Engineering, QC, Validation, Supply Chain, Warehouse, IT, and Regulatory teams may have limited involvement.
Quality issues rarely originate from a single department.
Therefore, sustainable solutions require multidisciplinary perspectives.
When CAPA ownership remains isolated, root causes remain hidden.
G. Legacy QMS Systems
Many organizations still rely on:
- Excel spreadsheets
- Manual trackers
- Email-based approvals
- Fragmented databases
These systems create challenges such as:
- Poor visibility
- Delayed escalation
- Limited trend analysis
- Lack of predictive insights
Modern quality management requires integrated digital ecosystems rather than disconnected administrative tools.
What Global Regulators Are Seeing
Regulatory authorities consistently identify CAPA deficiencies among the most frequent inspection observations.
US FDA Expectations
The FDA expects CAPA systems to:
- Identify quality issues
- Determine true root causes
- Implement effective corrections
- Verify long-term effectiveness
FDA Warning Letters frequently cite:
- Repeated deviations
- Weak investigations
- Unsupported root causes
- Ineffective CAPAs
EMA Expectations
The European Medicines Agency emphasizes:
- Quality risk management
- Knowledge management
- Continuous improvement
Repeated deviations often signal weaknesses in pharmaceutical quality systems.
MHRA Observations
The MHRA regularly identifies:
- Superficial investigations
- Weak effectiveness checks
- Recurring GMP failures
Inspectors increasingly expect organizations to demonstrate sustainable improvements.
WHO and PIC/S Expectations
WHO GMP and PIC/S guidance emphasize:
- Systematic investigations
- Risk-based decision making
- Data-driven quality management
- Continuous process improvement
Organizations unable to demonstrate CAPA effectiveness face heightened regulatory scrutiny.
The Cost of Ineffective CAPA
Many organizations underestimate the true cost of recurring deviations.
Product Quality Impact
Recurring failures increase the likelihood of:
- Batch rejection
- Product defects
- Process variability
- Market complaints
Patient Safety Risks
Every unresolved quality issue introduces potential risk to patients.
The pharmaceutical industry’s ultimate responsibility remains patient protection.
Regulatory Compliance Consequences
Ineffective CAPA systems can contribute to:
- Form 483 observations
- Warning Letters
- Import alerts
- Regulatory restrictions
Operational Inefficiency
Repeated investigations consume enormous resources.
Teams repeatedly spend time:
- Investigating
- Documenting
- Retraining
- Reviewing
without eliminating the underlying problem.
Manufacturing Productivity Losses
Recurring deviations contribute to:
- Downtime
- Delays
- Scrap
- Rework
- Capacity losses
Audit Readiness Challenges
Auditors routinely examine repeat observations.
Repeated deviations immediately raise concerns regarding CAPA effectiveness.
Reputation Damage
Customers, regulators, and stakeholders lose confidence when the same issues continue to reappear.
Financial Impact
The cumulative cost includes:
- Investigation expenses
- Product losses
- Compliance costs
- Delayed product releases
- Lost business opportunities
Poor CAPA performance directly affects profitability.
What Modern CAPA Systems Should Look Like
The pharmaceutical industry must evolve beyond traditional CAPA practices.
Intelligent Root Cause Analysis
Modern investigations should leverage structured methodologies including:
5 Why Analysis
Progressively explores underlying causes.
Fishbone Analysis
Identifies contributing factors across:
- People
- Process
- Equipment
- Materials
- Environment
- Measurement
Fault Tree Analysis
Maps failure pathways systematically.
Human Factors Engineering
Evaluates:
- Workload
- Ergonomics
- Process complexity
- User interactions
This approach shifts focus from blaming people to improving systems.
Risk-Based CAPA Prioritization
Organizations should prioritize CAPAs according to:
- Patient impact
- Product quality risk
- Compliance risk
- Business impact
Risk-based approaches ensure resources focus on the most critical vulnerabilities.
Data-Driven Decision Making
Future CAPA systems must utilize:
- Deviation trending
- Statistical analysis
- Quality metrics
- Process capability indicators
- Predictive quality models
Data should drive decisions—not assumptions.
Integration Across Quality Systems
CAPA should not operate independently.
It should integrate seamlessly with:
- Deviation Management
- Change Control
- Risk Management
- Training Systems
- Knowledge Management
- Supplier Quality Systems
Integration improves organizational learning and prevents quality silos.
Digital QMS Transformation
Modern CAPA systems increasingly utilize:
- Automated workflows
- Real-time dashboards
- Electronic investigations
- Risk scoring engines
- AI-supported analysis
Digital quality ecosystems improve visibility, consistency, and decision quality.
CAPA Effectiveness Framework
Organizations seeking sustainable improvement should adopt the following framework:
Step 1: Detect
Identify quality signals through:
- Deviations
- Complaints
- OOS events
- Audits
- Trending data
Step 2: Investigate
Conduct objective, data-driven investigations.
Avoid assumptions.
Follow evidence.
Step 3: Identify the True Root Cause
Challenge superficial conclusions.
Look beyond human error.
Focus on system vulnerabilities.
Step 4: Implement Sustainable Actions
Corrective actions should:
- Eliminate causes
- Reduce risk
- Strengthen controls
- Improve process robustness
Step 5: Verify Effectiveness
Measure outcomes rather than activities.
Evaluate whether recurrence risk has genuinely decreased.
Step 6: Monitor Trends
Continuously monitor:
- Recurrence rates
- Quality metrics
- Process performance
Trend analysis should become a routine management activity.
Step 7: Drive Continuous Improvement
Every CAPA should generate organizational learning.
The objective is not simply preventing recurrence but continuously improving quality systems.
Future of CAPA in Pharma
The future of pharmaceutical quality management will be shaped by Quality 4.0 principles.
AI-Powered Investigations
Artificial intelligence can assist in:
- Pattern recognition
- Trend identification
- Root cause hypothesis generation
- Investigation support
Predictive Quality Systems
Rather than reacting to deviations, organizations will increasingly predict them before occurrence.
Predictive analytics can identify emerging risks and initiate preventive actions proactively.
Digital Quality Management
Integrated digital platforms will provide:
- End-to-end visibility
- Real-time risk monitoring
- Automated escalation
- Continuous effectiveness tracking
Data-Driven Compliance Culture
The future organization will replace compliance-by-documentation with compliance-by-performance.
Quality decisions will be driven by:
- Evidence
- Analytics
- Risk intelligence
- Continuous learning
Key Takeaways
The pharmaceutical industry is facing a critical challenge:
Despite increasingly sophisticated quality systems, recurring deviations remain widespread.
The reason is clear.
Many CAPA programs focus on closure rather than effectiveness.
They address symptoms rather than root causes.
They emphasize documentation rather than improvement.
They measure activity rather than outcomes.
Recurring deviations are rarely isolated events.
They are warning signals that deeper system weaknesses remain unresolved.
Organizations that continue relying on traditional CAPA approaches will struggle with recurring quality failures, regulatory scrutiny, operational inefficiencies, and increasing compliance risks.
Organizations that embrace intelligent investigations, risk-based thinking, digital quality systems, predictive analytics, and continuous improvement will transform CAPA into a strategic business advantage.
The future of pharmaceutical quality does not belong to companies that close CAPAs fastest.
It belongs to companies that solve problems permanently.
Conclusion
CAPA remains one of the most powerful tools within the Pharmaceutical Quality System.
However, its effectiveness depends on how organizations use it.
When CAPA becomes a paperwork exercise, deviations repeat.
When CAPA becomes a system-improvement engine, quality improves.
The pharmaceutical industry must move beyond compliance-driven CAPA programs and adopt data-driven, risk-based, technology-enabled approaches that eliminate root causes and strengthen operational excellence.
Because ultimately, CAPA is not about investigations.
It is about learning.
It is about improvement.
And most importantly, it is about protecting patients.
“If the same deviation keeps returning, the problem was never truly solved. Effective CAPA is not about closing records—it is about eliminating risk, strengthening systems, and protecting patients.”